This information provides new insights into autoimmune mechanisms in general and could help develop and screen treatments for patients suffering from all autoimmune diseases, estimated to affect 5-10 percent of the U.S. population.
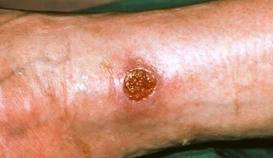
The study of pemphigoid rashes is of interest to people like me, who get these types of rashes-here's a pic of me with one-
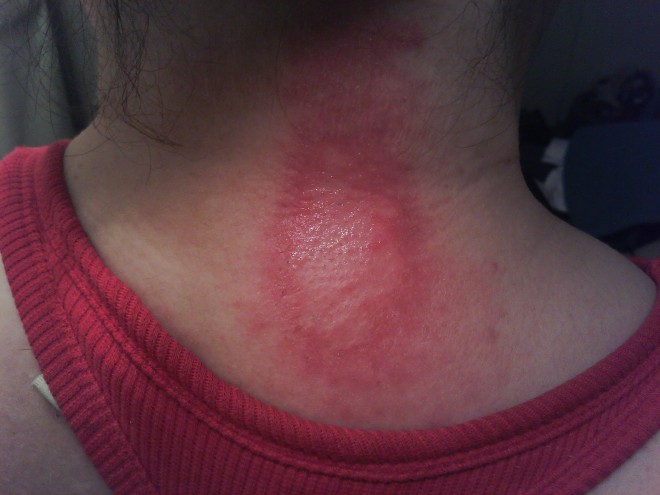
- My Pemphigoid Rash

I've only had one pemphigoid type rash since, and it was a small one. Since being treated for lupus with Cellcept (mycophenalate) my rashes (discoid, pemphigoid, porphyria, urticaria) have all been minimal. Much less severe!!! Of course now I stay in as much as possible, it's the only real way to minimize the rashes. Don't go out during the day!
Here's some info on Pemphigoid Rashes!
Definition
Pemphigoid is a group of subepidermal, blistering autoimmune diseases that primarily affect the skin, especially the lower abdomen, groin, and flexor surfaces of the extremities. Here, autoantibodies (anti-BPA-2 and anti-BPA-1) are directed against the basal layer of the epidermis and mucosa.The condition tends to persist for months or years with periods of exacerbation and remission. Localized variants of the condition have been reported, most often limited to the lower extremities and usually affecting women.
Types
There are two predominant types of pemphigoid: mucous membrane pemphigoid (MMP) also called cicatricial pemphigoid, and bullous pemphigoid (BP) (g). Pathogenesis and management are quite different for these conditions. Scar formation in mucous membrane pemphigoid can lead to major disability (g).Pemphigoid gestationis
Pemphigoid gestationis (PG) is a rare autoimmune bullous dermatosis of pregnancy. The disease was originally named herpes gestationis on the basis of the morphological herpetiform feature of the blisters, but this term is a misnomer because PG is not related to or associated with any active or prior herpes virus infection (h). PG typically manifests during late pregnancy, with an abrupt onset of extremely pruritic urticarial papules and blisters on the abdomen and trunk, but lesions may appear any time during pregnancy, and dramatic flares can occur at or immediately after delivery. PG usually resolves spontaneously within weeks to months after delivery.Epidemiology
Bullous Pemphigoid:
- BP is the most frequent blistering disease of the skin (and mucosa) affecting typically the elderly (>65 years) (k), but can occur at any age and in any race.
- Overall incidence: ± 7-10 new cases per million inhabitants per year.
- After the age of 70 incidence significantly increases.
- Relative risk for patients > 90 y have a 300-fold higher than those < 60 y.
- Women and men equally afflicted.
- Precipitating factors include trauma, burns, ionizing radiation, ultraviolet light, and certain drugs such as neuroleptics and diuretics, particularly lasix (furosemide), thiazides, and aldosterone antagonists.
- Correlations between BP flare activity and recurrence of underlying cancer suggest such an association in some patients.
- Even without therapy, BP can be self-limited, resolving after a period of many months to years, but is still a serious condition especially in the elderly.
- (j) 1-y survival probability may be as high as 88.96% (standard error 5.21%), with a 95% confidence interval (75.6%, 94.2%) but other analyses have documented 1 year mortalities of as much as 25-30% in moderate to severe pemphigus even on therapy (hh).
- Genetics
- Genetic predisposition, but not hereditary
Pemphigoid Gestationis:
- Is a condition of pregnancy (childbearing age women).
- In the United States, PG has an estimated prevalence of 1 case in 50,000-60,000 pregnancies.
- No increase in fetal or maternal mortality has been demonstrated, although a greater prevalence of premature and small-for-gestational-age (SGA) babies is associated with PG.
- Patients with PG have a higher relative prevalence of other autoimmune diseases, including Hashimoto thyroiditis, Graves disease, and pernicious anemia.
Histology
The earliest lesion of BP is a blister arising in the lamina lucida, between the basal membrane of keratinocytes and the lamina densa. This is associated with loss of anchoring filaments and hemidesmosomes. Histologically, there is a superficial inflammatory cell infiltrate and a subepidermal blister without necrotic keratinocytes. The infiltrate consists of lymphocytes and histiocytes and is particularly rich in eosinophils. There is no scarring.Approximately 70% to 80% of patients with active BP have circulating antibodies to one or more basement membrane zone antigens.
- Autoantibodies to BP180 (and BP230).
- BP180 and BP230 are two components of hemidesmosomes, junctional adhesion complexes.
- T cell autoreactive response to BP180 and BP230 regulate autoantibody production (l).
- On direct immunofluorescence, the antibodies are deposited in a thin linear pattern; and on immune electron microscopy, they are present in the lamina lucida. (By contrast, the antibodies to basement membrane zone antigens that are present in cutaneous lupus erythematosus are deposited in a granular pattern).
Clinical Features
Pemphigoid
Bullous Pemphigoid (BP) is subepidermal blistering autoimmune disease primarily affects the skin, especially the lower abdomen, groin, and flexor surfaces of the extremities. Mucous membrane involvement is seen in 10%-40% of patients. The disease tends to persist for months or years with periods of exacerbation and remission.The spectrum of presentations is extremely broad (l), but typically there is an itchy eruption with widespread blistering, and tense vesicles and bulla (blisters), with clear fluid (can be hemorrhagic) on apparently normal or slightly erythematous skin.
Lesions normally appear on the torso and flexures, particularly on the inner thighs. Blisters can range in size from a few millimeters to several centimeters, and although. pruritic, typically heal without scarring.
Sometimes erosions and crusting is seen. Also itchy bumps (papules) and crusts (plaques) can be seen with an annular or figurate pattern. A characteristic feature is that multiple bullae usually arise from large (palm-sized or larger), irregular, urticarial plaques. (r) Mucosal (oral, ocular, genital) involvement is also sometimes present, but ocular involvement, is rare. (s) BP can be difficult to diagnose in its ‘non-blistering’ stage, when just itchy, red, elevated patches are visible. Erosions are much less common than in pemphigus, and the Nikolsky sign is negative.
BP is characterized by spontaneous remissions followed by flares in disease activity that can persist for years. Even without therapy, BP is often self-limited, resolving after a period of many months to years, but may become very extensive.
Localized variants of the disease have been reported, most often limited to the lower extremities and usually affecting women. One such variant, localized vulvar pemphigoid, reported in girls aged 6 months to 8 years, presents with recurrent vulvar vesicles and ulcerations that do not result in scarring (t)
Bullous pemphigoid is distinguished from other blistering skin diseases, such as linear IgA dermatosis, epidermolysis bullosa acquisita, and cicatricial pemphigoid, by the following 4 clinical items (it can also be distinguished by biopsy and certain immunological tests) (u)
- Absence of atrophic scars;
- Absence of head and neck involvement;
- Relative absence of mucosal involvement.
Mucous membrane pemphigoid
Mucous membrane pemphigoid (MMP) is a chronic autoimmune disorder characterized by blistering lesions that primarily affect the various mucous membranes of the body, but also affects the skin (MMP is now the preferred term for lesions only involving the mucosa) (v). It is also known as Cicatricial Pemphigoid (CP), as it is often scarring.The mucous membranes of the mouth and eyes are most often affected, but those of the nose, throat, genitalia, and anus may also be affected. The symptoms of MMP vary among affected individuals depending upon the specific site(s) involved and the progression of the disease. Disease onset is usually between 40 and 70 years and oral lesions are seen as the initial manifestation of the disease in about two thirds of the cases. Blistering lesions eventually heal, sometimes with scarring. Progressive scarring may potentially lead to serious complications affecting the eyes and throat.
There is no racial or ethnic predilection although most studies have demonstrated a female to male ratio of approximately 2:1 (w). The diagnosis of MMP is mainly based on history, clinical examination and biopsy of the lesions.
MORE HERE from the University of Buffalo-